|
Ep 31 of Medtech Snapshot features Krystal Santiago sharing with us strategies for working with regulatory agencies during a submission and how utilizing an intermediary can be beneficial. This impacts decision making with clinical studies, product codes and requirements, pre-submission planning, etc. Did you miss the last 30 episodes of Medtech Snapshot? If so, check out our archives at https://www.sqr1services.com/white-papers/category/snapshot #medtechsnapshot #snapshot #medtech #medicaldevice #FDA #regulatory #productsubmission #compliance #510k #PMA 
0 Comments
Medtech Snapshot Part 2:2 continues the discussion as Trisha Aure covers considerations when transitioning from 21 CFR 820 to ISO 13485.
Hear how due diligence and training play a crucial role in the transition process and the impact it has on our employees. #iso13485 #regulation #compliance #fda #21CFR #riskmanagement #medicaldevice #medtech #news #podcast #snapshot #regulatoryaffairs In this 2-part series of Medtech Snapshot we're joined by Square-1 Engineering Director of Delivery & Operations Trisha Aure as she walks us through the highlights of our article 'FDA Announcement: 21 CFR 820 and ISO 13485 Guidance'
Hear the areas where 21 CFR 820 differs most from ISO 13485 and what this means for medical device OEMs. Part-2 of this series will cover the process to transition and key considerations when doing so. #iso13485 #regulation #compliance #fda #21CFR #riskmanagement #medicaldevice #medtech #news #podcast #snapshot #regulatoryaffairs This episode of Medtech Snapshot features our good friend Jeff Gable, medtech embedded software SME, diving in on Agile and NPD activities. Jeff walks us through his strategy for Agile development with FDA design controls, specifically within the design input phase. Hear Jeff's 4 step approach and how a tight feedback loop is critical between requirements and system design/ architectures. #agile #embeddedsoftware #productdevelopment #medicaldevice #medtech #NPD #FDA #designcontrol #designinput #snapshot  On February 2nd, 2024 the U.S. Food and Drug Administration (FDA) made its formal announcement and ruling providing guidance on 21 CFR Part 820 which up to that point was the standard overseeing medical device quality system regulation and current good manufacturing practices (cGMP) in the United States of America. The big news – 21 CFR 820 will be heavily amended to incorporate ISO 13485 as the leading guidance for Quality Management System Regulation (QMSR) and cGMP. While the news wasn’t a surprise, it does put a final note on the direction the agency intends to take for medical device practices moving forward, especially as it relates to risk management. While many device OEMs already utilize ISO as their leading regulation standard, those who don’t will have two years to adjust to these changes to be in compliance effective February 2nd, 2026. Here’s what you need to know as it relates to the differences between 21 CFR 820 and ISO 13485, as well as considerations OEMs should take into account in order to meet the 2026 deadline. Big Picture:
Key Differences: 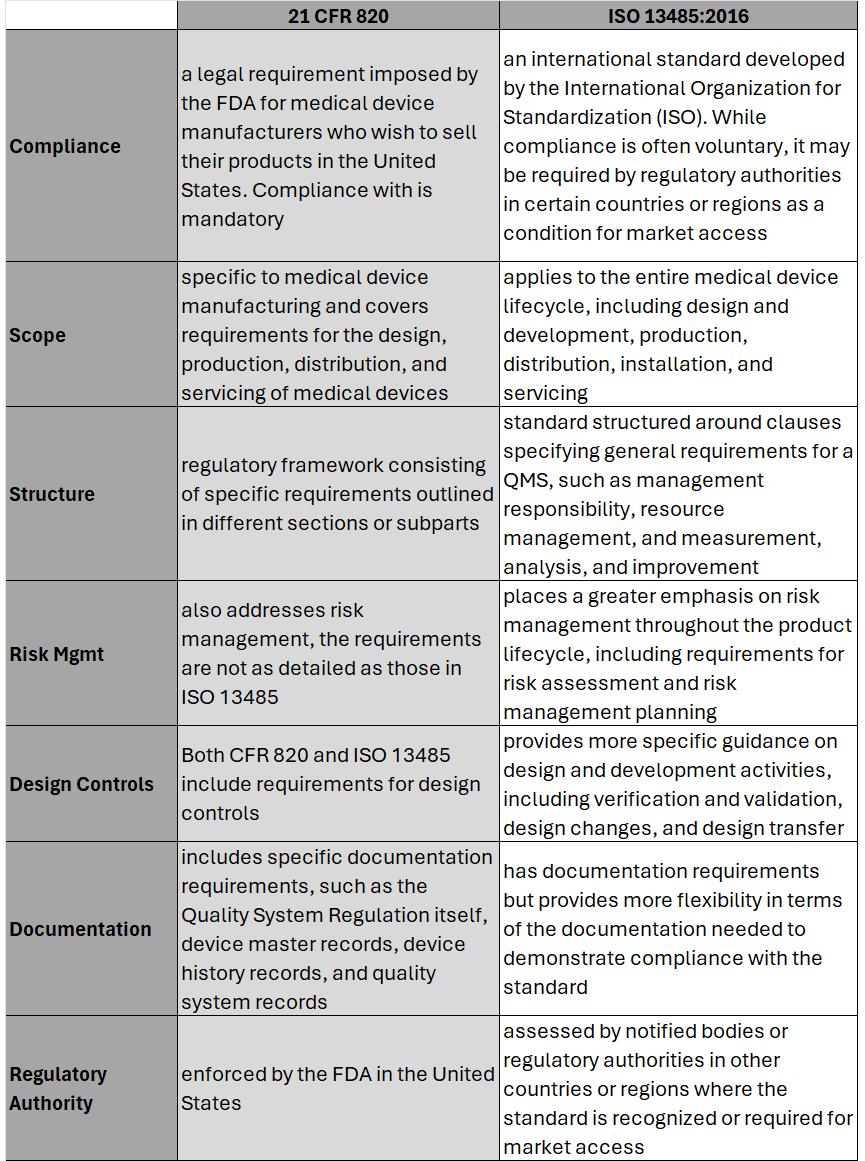 Transitioning to ISO 13485 Transitioning from CFR 820 to ISO 13485 involves several steps to ensure compliance with the ISO standard. A general outline of the steps an OEM should take to transition may include: 1. Understand the Requirements of ISO 13485: Familiarize yourself with the requirements of ISO 13485. This includes understanding the structure of the standard, its key clauses, and any specific requirements which may differ from CFR 820. (see above table for highlights) 2. Gap Analysis: Conduct a thorough gap analysis to identify the differences between your current quality management system under CFR 820 and the requirements of ISO 13485. This will help you determine what changes, if any, need to be made to your existing processes, procedures, and documentation. This is also a great time to do a review of your QMS tool to determine if it is an appropriate tool for future use. 3. Document Review and Update: Review your existing documentation, including quality manuals, procedures, work instructions, and forms, to ensure they align with the requirements of ISO 13485. Update or create new documents as necessary to meet the standard's requirements. 4. Training and Awareness: Provide training to relevant personnel to ensure they understand the requirements of ISO 13485 and their roles in implementing and maintaining the QMS. This may include training on new procedures, processes, and documentation. 5. Implementation of New Processes: Implement any new processes or procedures required by ISO 13485. This may include processes related to risk management, design and development, purchasing, production, and service provisions. 6. Internal Audits: Conduct internal audits of your QMS to verify compliance with ISO 13485 requirements. Identify any non-conformities and take corrective actions to address them. 7. Management Review: Hold management reviews to evaluate the effectiveness of the QMS and identify opportunities for improvement. Ensure top management is actively involved in the transition process and committed to maintaining the QMS. 8. Certification Audit: BEFORE you consider this step be sure to speak with a regulatory affairs subject matter expert to ensure it is necessary. Once you believe your QMS is fully compliant with ISO 13485, engage a certification body to conduct a certification audit. The audit will assess your organization's compliance with the standard and determine if you are eligible for certification. 9. Address Non-conformities: If any non-conformities are identified during the certification audit, take corrective actions to address them. The certification body will typically require verification that corrective actions have been implemented before issuing the ISO 13485 certificate. 10. Continual Improvement: Continuously monitor and improve your QMS to ensure ongoing compliance with ISO 13485 and to enhance the efficiency and effectiveness of your processes. Although the 21 CFR 820 and ISO 13485 vary in their structure, and at times use different terminology to describe similar concepts, 21 CFR 820 and ISO 13485 are substantially similar in that both prioritize principles such as risk management, design controls, and continual process improvement. It’s possible as organizations begin to look at their current standards and systems, they will find the transition process is not as cumbersome as initially thought. While this is an obvious assumption, it’s important to note regulatory affairs professionals should be counseled throughout this entire process to ensure appropriateness of adoption and change management.  Medical device companies play a critical role in advancing healthcare as their ability to diagnose, monitor, and treat medical conditions allow patients like you and I the opportunity to recover and live longer. Device companies carry a heavy burden on our behalf and that burden starts with product risk. One of the biggest challenges an OEM medical device organization will be faced with is managing risk, especially during the early stages of product development. The integration of risk management into design control (ISO 14971) is essential for identifying, assessing, and mitigating potential risks associated with the design and development of a medical device. Given risk management is a part of nearly every development process, and is a primary focus of all regulatory agencies like the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA), why is it then so many medical device companies struggle with sound risk management strategies? The failure to address risks adequately can lead to a whole host of problems ranging from regulatory non-compliance, compromised patient safety, financial setbacks, and in severe situations criminal prosecution of executives. Needless to say, understanding why medical device companies come up short with their risk management strategy and how you can avoid that for yourself is key to your future success. In this article, we will explore some of the key reasons behind risk management failure. Most Common Risk Failure Factor - Inadequate Understanding of Regulatory Requirements: One of the primary reasons for failure in risk management is an insufficient understanding of the complex and evolving regulatory landscape. Medical device companies must navigate a web of regulations, standards, and guidelines to ensure compliance. Failing to keep abreast of these requirements can result in flawed risk assessments, inadequate risk mitigation measures, and ultimately, regulatory sanctions. Poor Integration of Risk Management into Product Development: Successful risk management should be an integral part of the product development lifecycle. However, some companies make the mistake of treating it as a standalone process rather than integrating it seamlessly into every stage of development. When risk management is an afterthought, essential risks may be overlooked, leading to suboptimal product designs, increased failure rates, and compromised patient outcomes. Lack of Cross-Functional Collaboration: Effective risk management requires collaboration across various departments, including research and development, regulatory affairs, quality assurance, and manufacturing. Failure to establish clear communication channels and encourage cross-functional collaboration can result in siloed decision-making. This lack of coordination can lead to critical risks being underestimated or missed entirely. Insufficient Resources and Expertise: Some medical device companies fail in risk management due to resource constraints and a shortage of expertise. This can manifest in inadequate training for personnel responsible for risk management, insufficient allocation of time and budget, and a lack of access to external expertise. Without the necessary resources, companies may struggle to conduct comprehensive risk assessments and implement effective risk mitigation strategies. Overemphasis on Short-Term Goals: Pressure to meet short-term financial goals can sometimes lead companies to prioritize speed to market over thorough risk analysis. This can result in hasty decision-making and inadequate risk identification and mitigation. Companies need to strike a balance between meeting market demands and ensuring the safety and efficacy of their medical devices in the long run. Failure to Learn from Industry Incidents: The medical device industry has witnessed several high-profile incidents related to product failures and patient harm. Failure to learn from these incidents and implement lessons learned into future risk management strategies can perpetuate the same mistakes. Companies must actively analyze industry incidents, update risk management processes accordingly, and continuously improve their practices. Ineffective Communication with Stakeholders: Communication is crucial in risk management, both internally and externally. Companies that fail to communicate effectively with their stakeholders, including regulatory bodies, healthcare professionals, and patients, may face increased scrutiny and regulatory challenges. Transparency and open communication are essential for building trust and demonstrating commitment to patient safety. In the highly regulated and dynamic field of medical devices, effective risk management is not just a regulatory requirement - it is a fundamental aspect of ensuring patient safety and the success of a company. Understanding the pitfalls that lead to failures in risk management can help medical device companies proactively address these challenges. By prioritizing compliance, integrating risk management into every stage of product development, fostering cross-functional collaboration, and learning from industry incidents, companies can enhance their risk management strategies and contribute to the advancement of healthcare with safe and reliable medical devices. The quickest way to overcome a business challenge is to get help from those who are experienced in besting your beast! The team at Square-1 Engineering is comprised of a variety of technical and project management professionals who are subject matter experts in the areas of NPD, Quality, Compliance and Manufacturing Engineering. Learn more about how we can solve your work and project problems today to get you back on track!
In this episode of Medtech Snapshot we're joined by SPARK Neuro COO Marinela Gombosev as we sort through the opportunities and challenges in adopting machine learning (ML) and artificial intelligence (AI) technologies in the medical device space. Listen in as Marinela provides additional perspective on the considerations for adopting this technology within our current health system and its stakeholders like the FDA, clinicians, etc.
Enjoying these quick 3-minute discussions? Be sure to check out our past Medtech Snapshot episodes at https://lnkd.in/gFwF9GYN #medtechsnapshot #medtech #snapshot #AI #machinelearning #medicaldevice #artificialintelligence #fda #healthcare  In most regulated industries remediation is a cost of doing business. Unfortunately the medical device industry is no different. While remediation won’t hit every business, the fact of the matter is as regulations continue to change and or grow more companies will find themselves in a spot where they are having to change their processes and procedures in order to remain in compliance. In 2021 we wrote about the keys to success, identifying six (6) key areas of focus to help one get through remediation and come out on the other end still in tact and moving forward. As the medical device industry continues to evolve, so must our approach to solving problems we face. As such, understanding the reasons why a device company may experience failure as they go through remediation is key to learning from others mistakes so we don’t repeat them when it comes our turn. WHY THINGS GO SIDEWAYS the top 11 reasons why remediation goes wrong for medical device OEMs:
When looking at this list the biggest take away is the starting point. Once it has been determined remediation is necessary, whether through regulatory intervention or internal, understanding what occurred to get us there in the first place is critical. If we misdiagnose the root cause of the problem within our operations our ability to successfully navigate through the rest of remediation is be hampered significantly. For this reason its wise to spend as much time as possible sorting through the cause and effects of your operation to accurately determine the root cause leading to remediation. Rushing this process will inevitably cause unnecessary challenges on the back end. SOLVING THE PROBLEM The quickest way to overcome a business challenge is to get help from those who are experienced in besting your beast! The team at Square-1 Engineering is comprised of a variety of technical and project management professionals who are subject matter experts in NPD, Quality, Compliance (and yes - remediation) and Manufacturing Engineering. Learn more about how we can solve your compliance problems while besting your remediation beast!  The results are in from our Square-1 Engineering online poll...."What are the top 2 most frequent reasons warning letters/ citations are issued by the FDA?"
Listen in as our managing director, Travis Smith, covers the results of he poll while sharing commentary from the medical device community. Need help dealing with an audit or warning letter? Learn more about our support capabilities at https://lnkd.in/g7NX_8pw #poll #fda #capa #complaints #regulatory #quality #medtech #square1engineering  Our client, a Class III medical device company, was issued an FDA 483 warning letter due to product field failures and QMS issues posing life threatening risks to implant patients. The Square-1 Engineering subject matter experts (SME team) were brought in to identify & correct systems and process issues across six (6) functional departments and two (2) manufacturing locations domestically. As you can guess our clients’ situation was dire due to several implant failures in the field. We invite you to download the case study below to learn how we saved our client $4.5M while successfully concluding the remediation project.
About the AuthorTravis Smith is the founder and managing director of Square-1 Engineering, a medical device consulting firm, providing end to end engineering and compliance services. He successfully served the life sciences marketplace in SoCal for over 15 years and has been recognized as a ‘40 Under 40’ honoree by the Greater Irvine Chamber of Commerce as a top leader in Orange County, CA. Categories
All
Archives
July 2024
|
||
 RSS Feed
RSS Feed
Visit Square-1's
|
|

