 On February 2nd, 2024 the U.S. Food and Drug Administration (FDA) made its formal announcement and ruling providing guidance on 21 CFR Part 820 which up to that point was the standard overseeing medical device quality system regulation and current good manufacturing practices (cGMP) in the United States of America. The big news – 21 CFR 820 will be heavily amended to incorporate ISO 13485 as the leading guidance for Quality Management System Regulation (QMSR) and cGMP. While the news wasn’t a surprise, it does put a final note on the direction the agency intends to take for medical device practices moving forward, especially as it relates to risk management. While many device OEMs already utilize ISO as their leading regulation standard, those who don’t will have two years to adjust to these changes to be in compliance effective February 2nd, 2026. Here’s what you need to know as it relates to the differences between 21 CFR 820 and ISO 13485, as well as considerations OEMs should take into account in order to meet the 2026 deadline. Big Picture:
Key Differences: 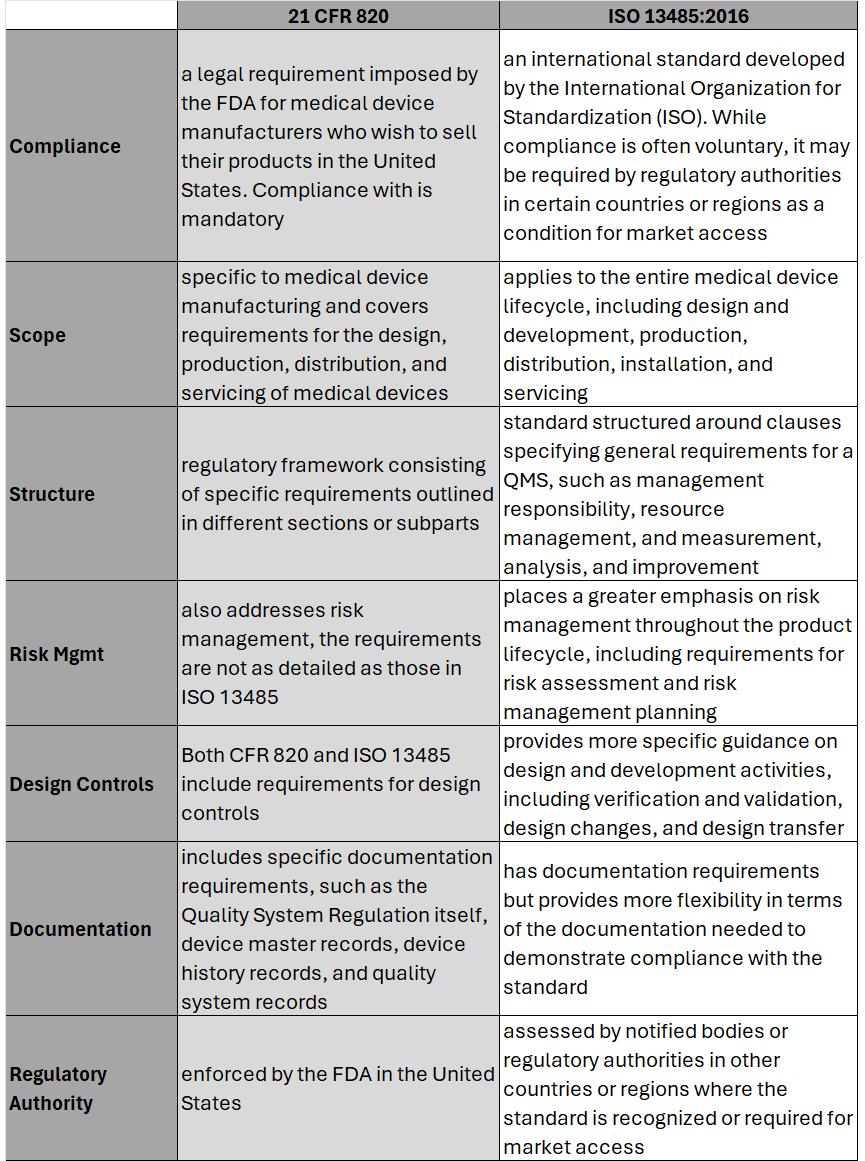 Transitioning to ISO 13485 Transitioning from CFR 820 to ISO 13485 involves several steps to ensure compliance with the ISO standard. A general outline of the steps an OEM should take to transition may include: 1. Understand the Requirements of ISO 13485: Familiarize yourself with the requirements of ISO 13485. This includes understanding the structure of the standard, its key clauses, and any specific requirements which may differ from CFR 820. (see above table for highlights) 2. Gap Analysis: Conduct a thorough gap analysis to identify the differences between your current quality management system under CFR 820 and the requirements of ISO 13485. This will help you determine what changes, if any, need to be made to your existing processes, procedures, and documentation. This is also a great time to do a review of your QMS tool to determine if it is an appropriate tool for future use. 3. Document Review and Update: Review your existing documentation, including quality manuals, procedures, work instructions, and forms, to ensure they align with the requirements of ISO 13485. Update or create new documents as necessary to meet the standard's requirements. 4. Training and Awareness: Provide training to relevant personnel to ensure they understand the requirements of ISO 13485 and their roles in implementing and maintaining the QMS. This may include training on new procedures, processes, and documentation. 5. Implementation of New Processes: Implement any new processes or procedures required by ISO 13485. This may include processes related to risk management, design and development, purchasing, production, and service provisions. 6. Internal Audits: Conduct internal audits of your QMS to verify compliance with ISO 13485 requirements. Identify any non-conformities and take corrective actions to address them. 7. Management Review: Hold management reviews to evaluate the effectiveness of the QMS and identify opportunities for improvement. Ensure top management is actively involved in the transition process and committed to maintaining the QMS. 8. Certification Audit: BEFORE you consider this step be sure to speak with a regulatory affairs subject matter expert to ensure it is necessary. Once you believe your QMS is fully compliant with ISO 13485, engage a certification body to conduct a certification audit. The audit will assess your organization's compliance with the standard and determine if you are eligible for certification. 9. Address Non-conformities: If any non-conformities are identified during the certification audit, take corrective actions to address them. The certification body will typically require verification that corrective actions have been implemented before issuing the ISO 13485 certificate. 10. Continual Improvement: Continuously monitor and improve your QMS to ensure ongoing compliance with ISO 13485 and to enhance the efficiency and effectiveness of your processes. Although the 21 CFR 820 and ISO 13485 vary in their structure, and at times use different terminology to describe similar concepts, 21 CFR 820 and ISO 13485 are substantially similar in that both prioritize principles such as risk management, design controls, and continual process improvement. It’s possible as organizations begin to look at their current standards and systems, they will find the transition process is not as cumbersome as initially thought. While this is an obvious assumption, it’s important to note regulatory affairs professionals should be counseled throughout this entire process to ensure appropriateness of adoption and change management. 
0 Comments
EU MDR/ IVDR PROPOSED PUSH1/25/2023  DID YOU KNOW On January 6, 2023 the European Commission, a political and regulatory steering committee consisting of a group of 27 Commissioners, known as 'the College', adopted a proposal to give more time to device OEMs to certify medical devices under EU MDR to mitigate the risk of shortages. The proposal, which now needs to be adopted by the European Parliament, could push out MDR requirements several years. Higher risk devices such as pacemakers and joint implants would have a shorter transition period till December 2027, whereas lower risk devices, such as syringes or reusable surgical instruments wouldn't be until December 2028. WHAT DOES THIS MEAN FOR YOU? Regardless of EU Parliament's decision to potentially extend MDR, device OEMs should consider the following as we hedge through 2023: 1. Strategies for US product approval and or commercialization will continue to increase as OEMs seek alternative pathways to potentially avoid EU MDR compliance. 2. As a result of #1, support to aide OEMs in their go-to-market strategy will intensify causing a shortage for resources, while potentially lengthening the process to get to approvals (supply & demand constraints - notified bodies and consulting firms experience increases in demand causing support shortages). This will be especially true with remediation work. 3. The idea of putting off or slowing MDR related efforts in the interim to re-focus on other activities may provide momentary relief, however it also creates a long-term liability in the business. This liability comes with a variety of future unknowns: regulatory landscape, inflation, cost of resources, CRO and notified body constraints, etc. If you must achieve MDR compliance our recommendation is to get it done and over with in the present. 4. Work associated with achieving MDR compliance can be easily underestimated, especially if you have legacy product where your CE mark was granted pre mid 2000s. The burden to meet MDR requirements may be steep, which is all the more reason to avoid procrastinating said efforts as outlined in #4.  SOLVING THE PROBLEM The quickest way to overcome a business challenge is to get help from those who are experienced in besting your beast! The team at Square-1 Engineering is comprised of a variety of technical and project management professionals who are subject matter experts in the areas of NPD, Quality, Compliance (and yes - remediation) and Manufacturing Engineering. Learn more about how we can solve your compliance problems while besting your EU MDR beast! Learn about Square-1 Engineering's mission and what it means to be fearless!  About the AuthorTravis Smith is the founder and managing director of Square-1 Engineering, a medical device consulting firm, providing end to end engineering and compliance services. He successfully served the life sciences marketplace in SoCal for over 15 years and has been recognized as a ‘40 Under 40’ honoree by the Greater Irvine Chamber of Commerce as a top leader in Orange County, CA. Categories
All
Archives
July 2024
|
 RSS Feed
RSS Feed
Visit Square-1's
|
|

